Table of Contents
1.1 The Compatibility of Browser and Operating System (OS)
druglikeFilter is free and open to all users with no login requirement and can be readily accessed by a variety of popular web browsers and operating systems as shown below.

1.2 Required Formats of the Input Files
druglikeFilter supports both SMILES (Simplified Molecular Input Line Entry System) and SDF (Structure Data File) formats. For convenience, SMILES strings can be consolidated into a TXT file (downloaded), and SDF files (downloaded) can include multiple compounds. It is recommended to upload no more than 10,000 molecules at a time. Molecules can be uploaded in three different ways:
1.2.1 Draw your own compound
Chemical structures can be drawn directly with the JMSE editor and then uploaded for further analysis. This provides a seamless workflow when creating and processing chemical structures.
1.2.2 Batch upload
Files in SMILES or SDF format can be selected and uploaded from your computer. An example file is provided to help you get started quickly, offering a convenient way to explore the server's functionality without preparing your own data initially.
Each molecule in the uploaded file will be automatically assigned a unique identifier based on the upload order (e.g., D0001, D0002). Detailed information about each molecule will be displayed, and you can click DETAIL to view additional properties for each compound.
1.2.3 Provide compound SMILES
SMILES can be uploaded by pasting the compound strings directly. An example is available to assist you in starting quickly, and a reset option allows you to clear the input. Each molecule in the uploaded file is automatically assigned a unique identifier based on the upload order (e.g., D0001, D0002). Detailed information about each molecule will be displayed, with the option to click DETAIL to view additional properties for each compound.

2. Evaluating physicochemical rule through systematic determination
druglikeFilter applies 12 drug-likeness rules and 15 commonly used physicochemical properties to identify and exclude non-druggable molecules.
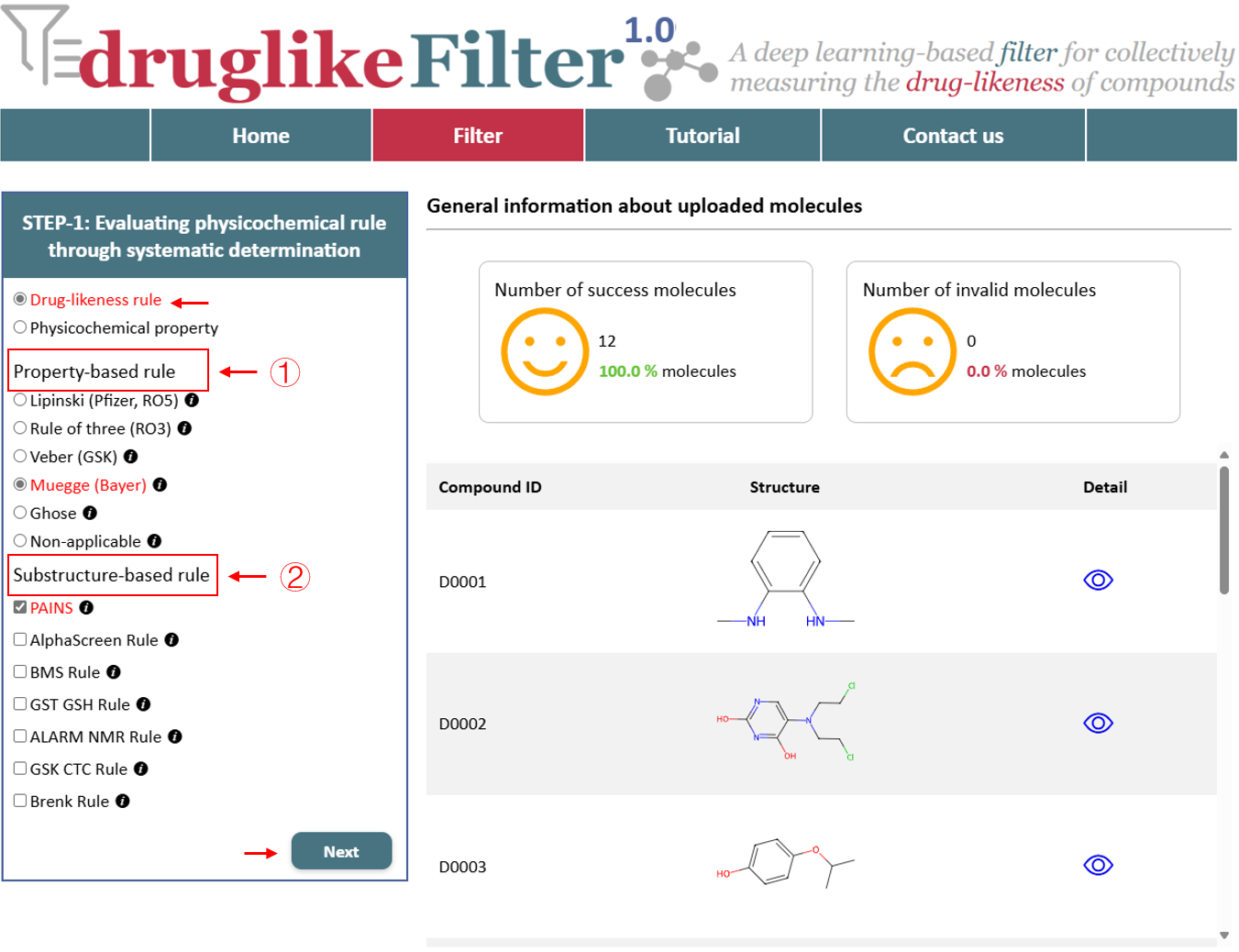
2.1 Drug-likeness rule
The drug-likeness evaluation consists of 5 property-based rules and 7 substructure-based rules. These rules help quickly eliminate non-drug-like molecules. However, different screening strategies are suited for different purposes. To accommodate various needs, users can choose the non-applicable option to skip this step entirely and proceed directly to the next stage without applying any filtering rules.
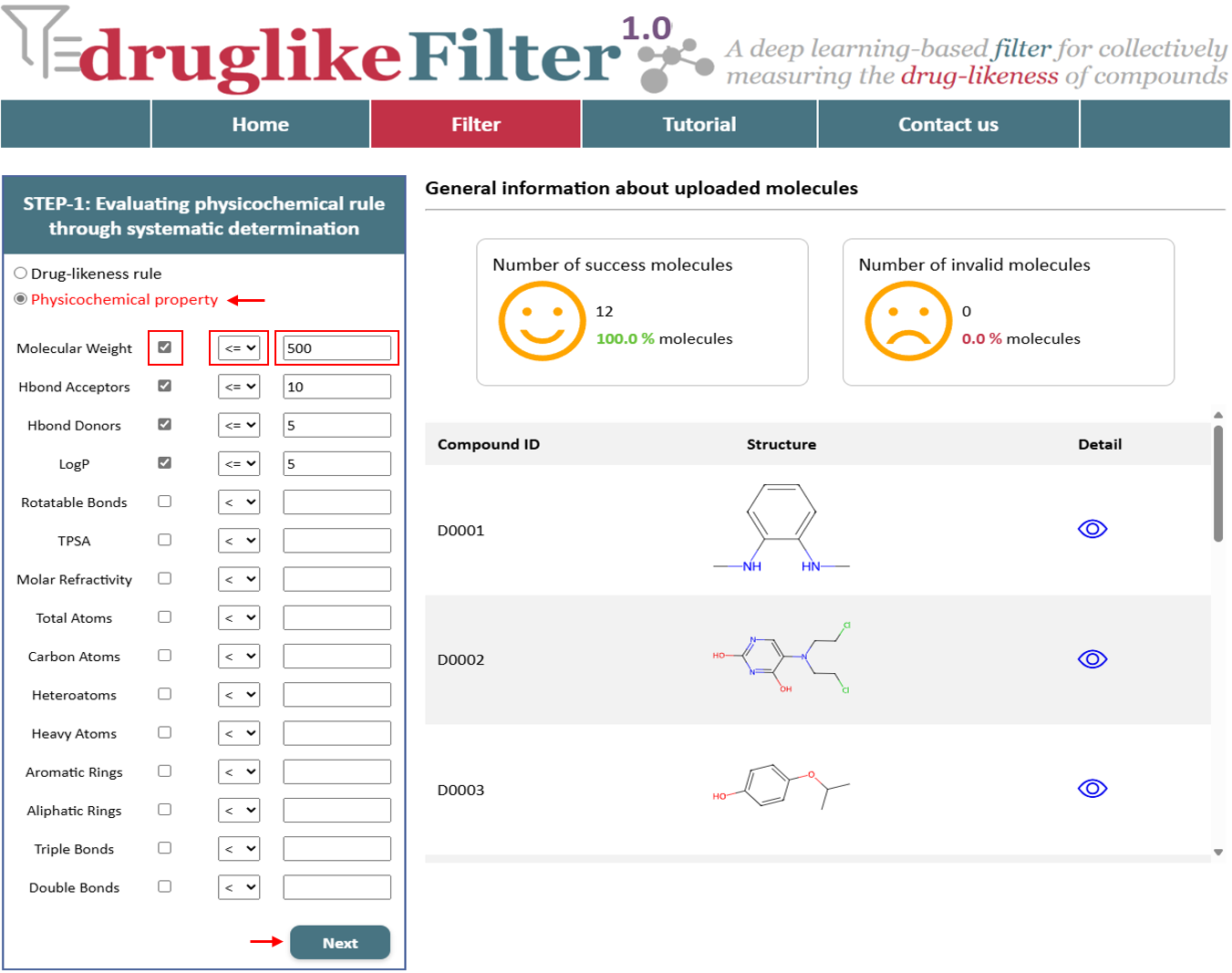
2.2 Physicochemical property
In general, screening strategies should be tailored to the specific objectives of each task. To accommodate various needs, druglikeFilter utilizes 15 commonly used physicochemical properties that can be used to flexibly customize compound screening hair schemes.
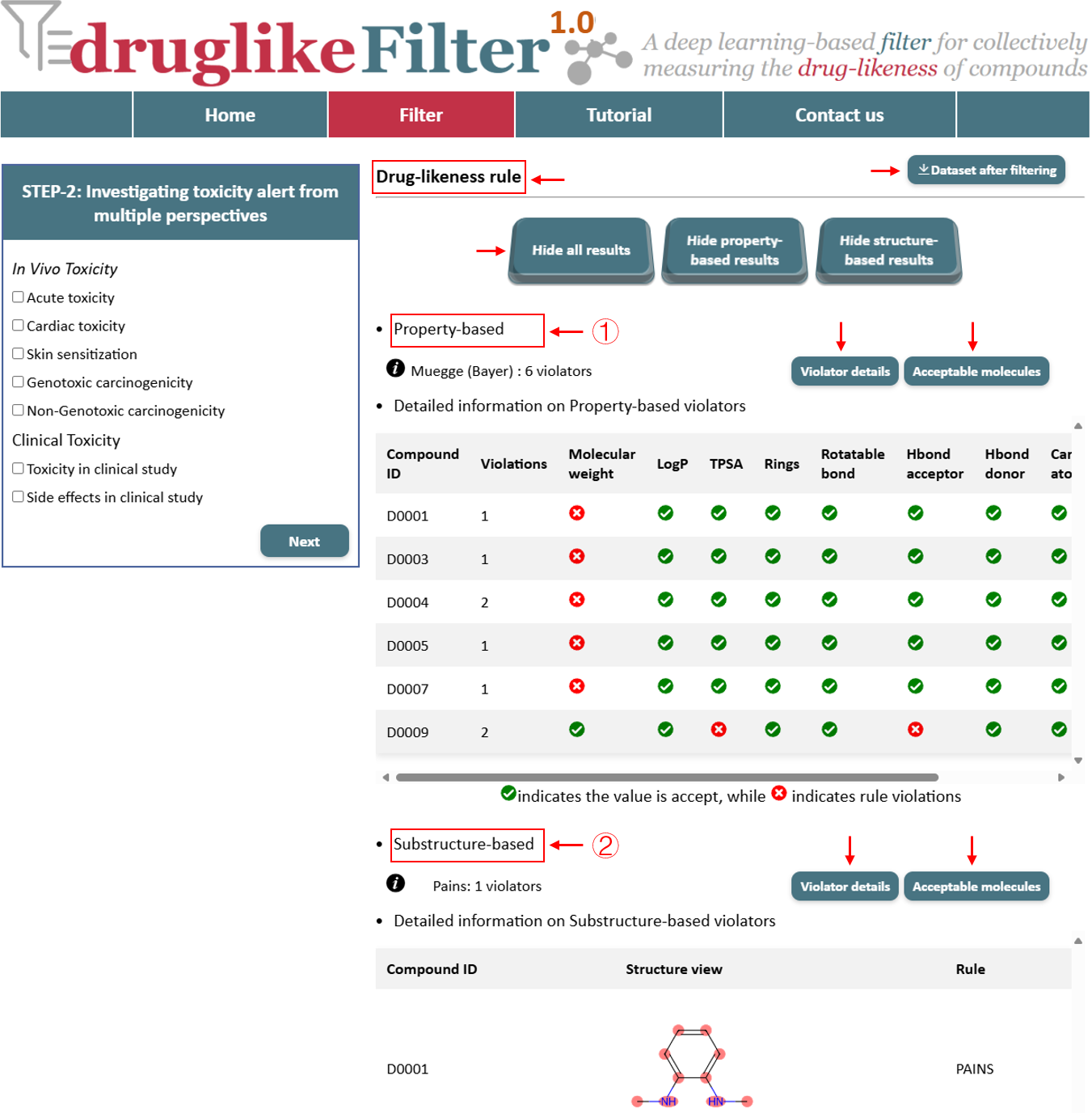
These results include property-based and substructure-based evaluations, with the number of violators and detailed information presented in a table. The calculated results are displayed separately, with options to show or hide them by clicking buttons. Compounds identified as drug-like are available for download at the top of the page and are automatically passed to the next step for further processing.
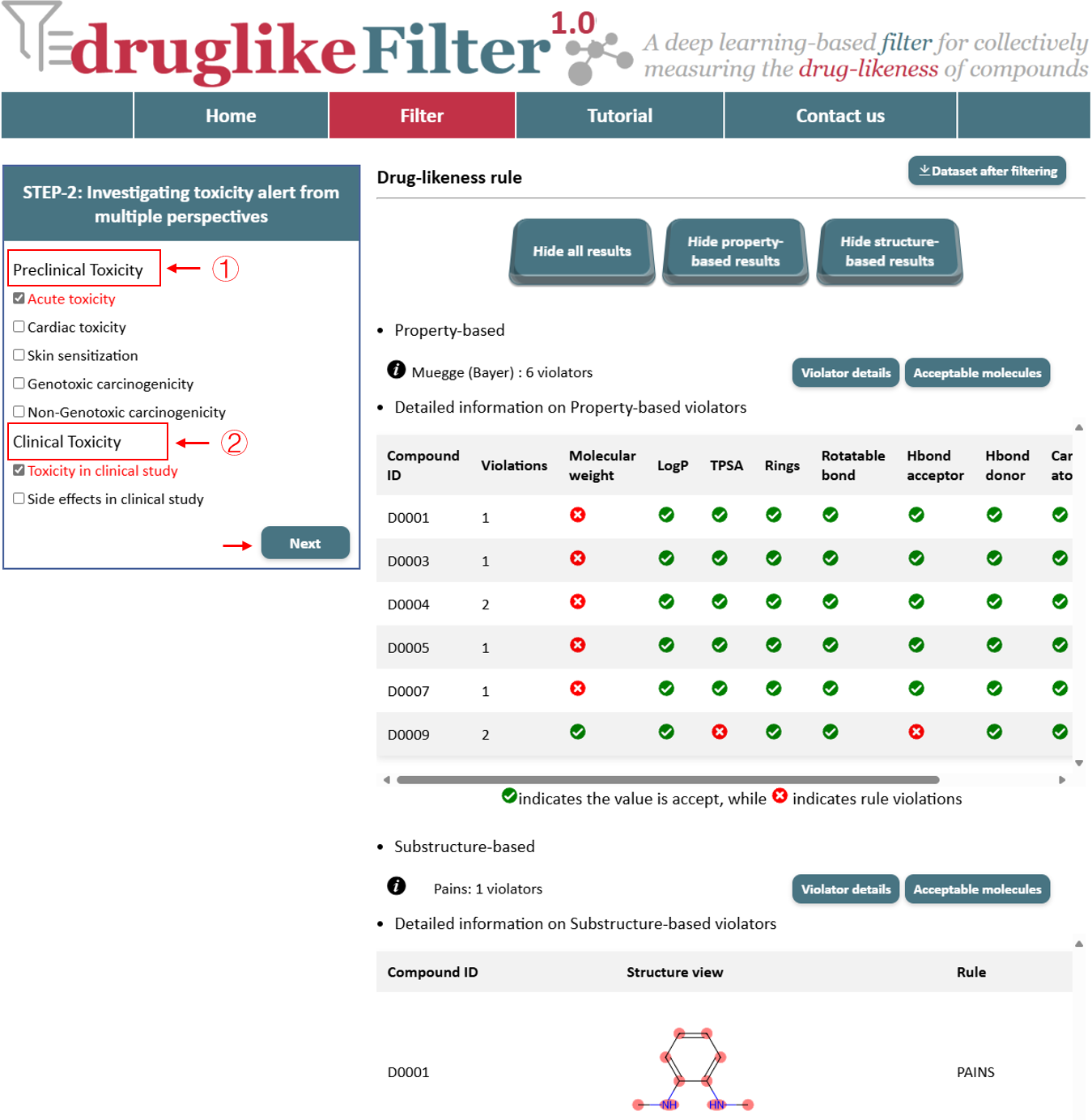
3. Investigating toxicity alert from multiple perspectives
druglikeFilter compiles approximately 600 toxicity alerts, derived from relevant preclinical and clinical studies, as well as deep learning model, to exclude molecules with potential toxicity risks.
3.1 Preclinical Toxicity
Toxicity alerts and deep-learning models is used to identify molecules with toxicological risks, including acute toxicity, skin sensitization, genotoxic carcinogenicity, non-genotoxic carcinogenicity, and others.
3.2 Clinical Toxicity
Toxicity alerts were derived from compounds in clinical studies and were used to exclude molecules at risk of toxicity. The calculated results include perclinical toxicity and clinical toxicity, with the number of violators and detailed information presented in a table. Compounds identified as drug-like are available for download at the top of the page and are automatically passed to the next step for further processing.
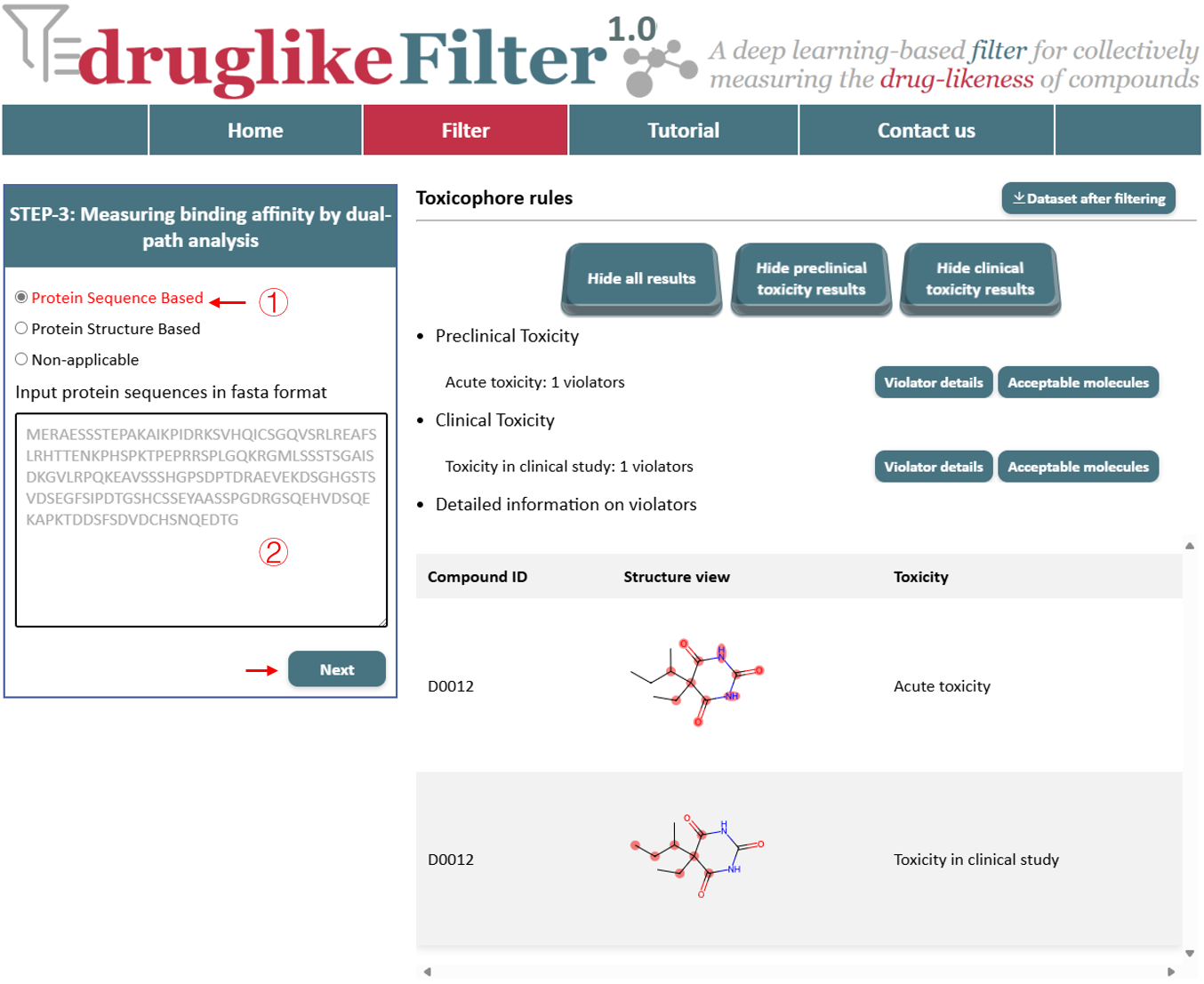
4. Measuring binding affinity used dual-path analysis
druglikeFilter offers two approaches to measure binding affinity: a structure-based method and a sequence-based method. Users can choose the method that best suits their data and research needs.
4.1 Protein Sequence Based
For drug targets with unavailable protein structure information, users can predict binding affinity by inputting the target sequence (FASTA) for binding affinity prediction. Click the "NEXT" button to proceed with binding affinity prediction. Top-ranked compounds will receive more stars, indicating greater binding affinity.
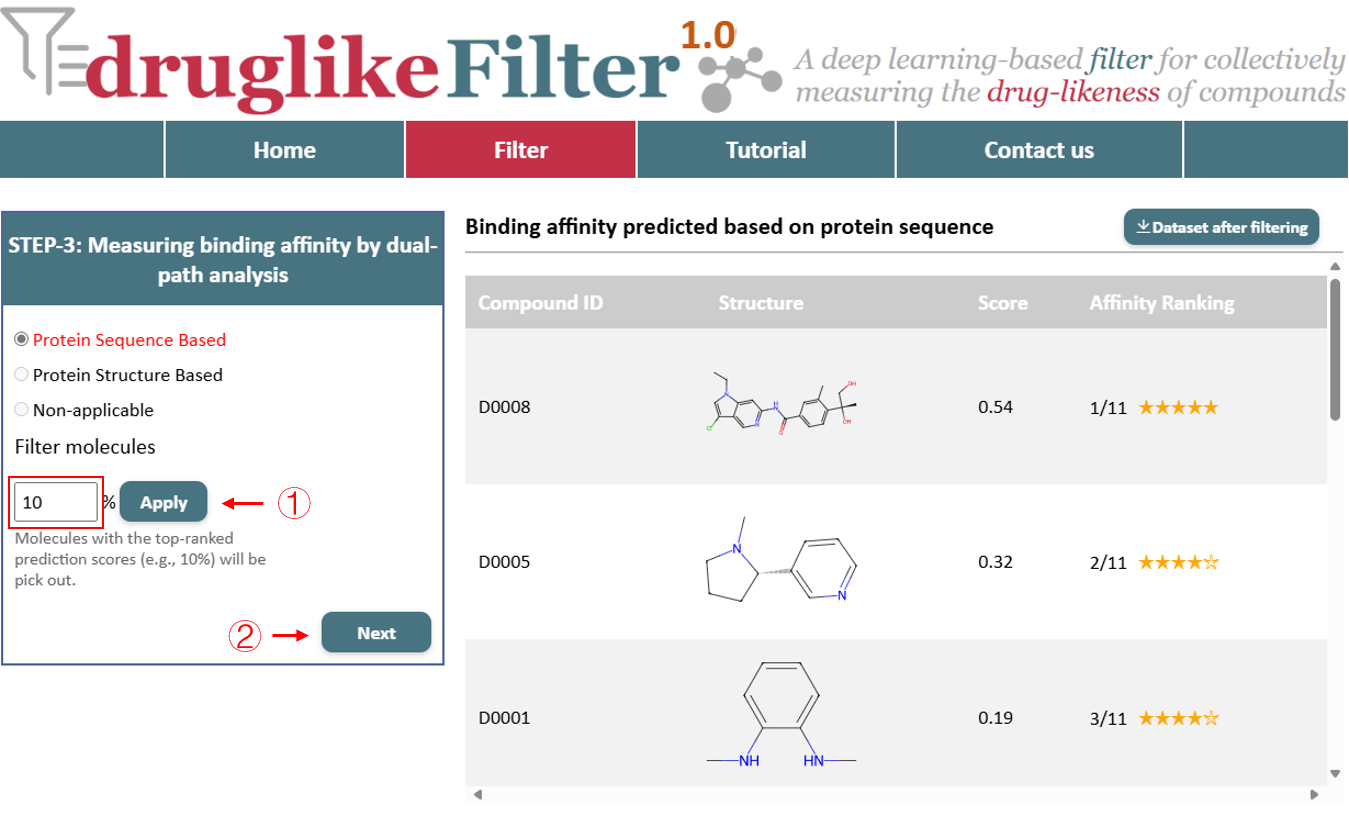
Additionally, the “Apply” button filters compounds based on the percentage value entered by the user, retaining those that meet the affinity criteria and dynamically updating the results table on the right. Click the "Dataset after filtering" button in the top-right corner for offline analysis, and these compounds will be passed to the next step for further processing.
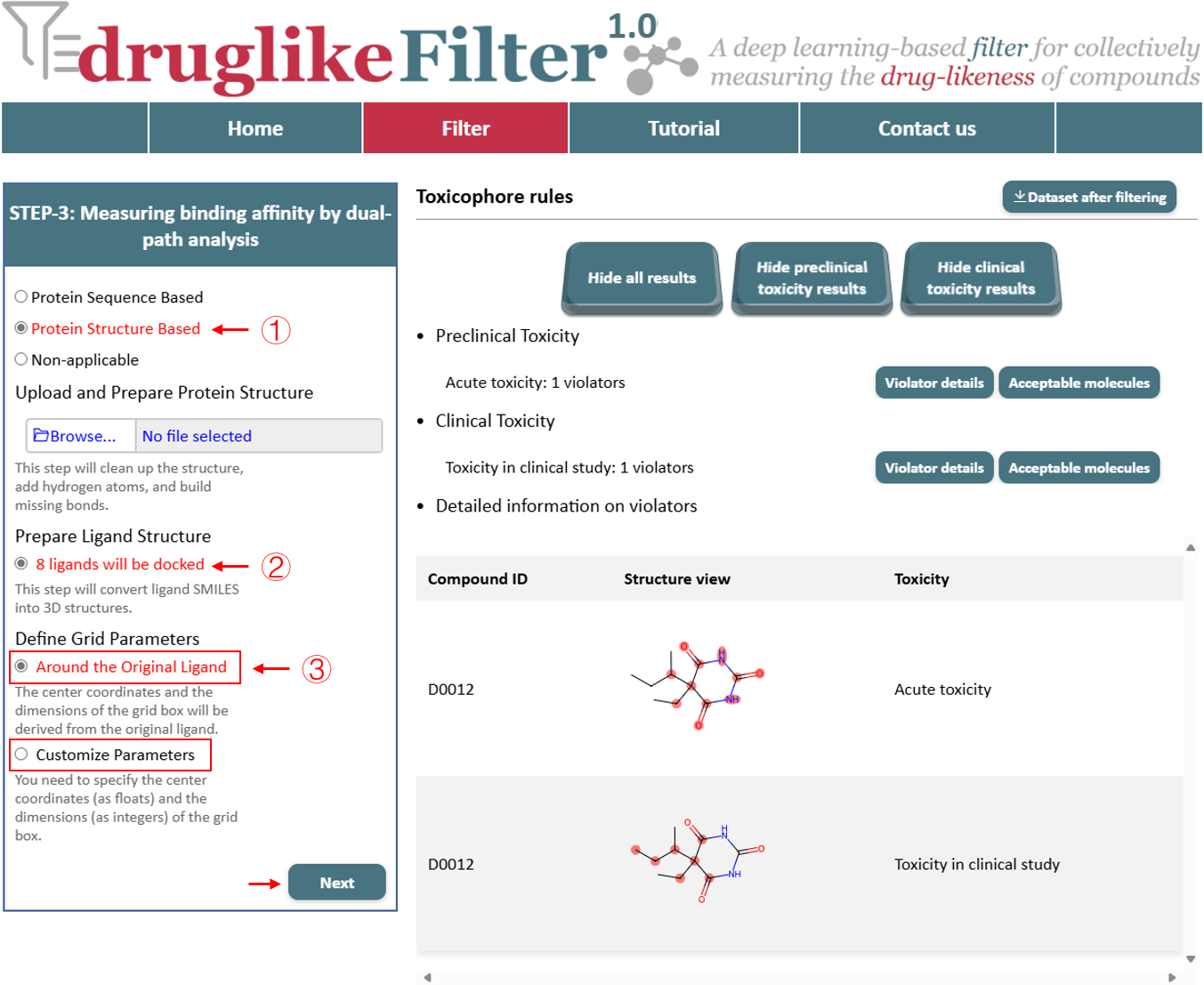
4.2 Protein Structure Based
When a protein structure file is uploaded, the system automatically cleans the structure, adds hydrogen atoms, and rebuilds any missing bonds. Ligands are converted into 3D structures and validated. By default, the binding pocket is defined around the original ligand, with the user selecting the ligand based on the protein structure information. Additionally, the binding pocket can be customized by specifying x, y, z coordinates to target a specific region. When using this method, users must provide the grid boundary size and the coordinates for the pocket center. The “Apply” button filters compounds based on the percentage value entered by the user, retaining those that meet the affinity criteria and dynamically updating the results table on the right. Click the "Dataset after filtering" button in the top-right corner for offline analysis, and these compounds will be passed to the next step for further processing.
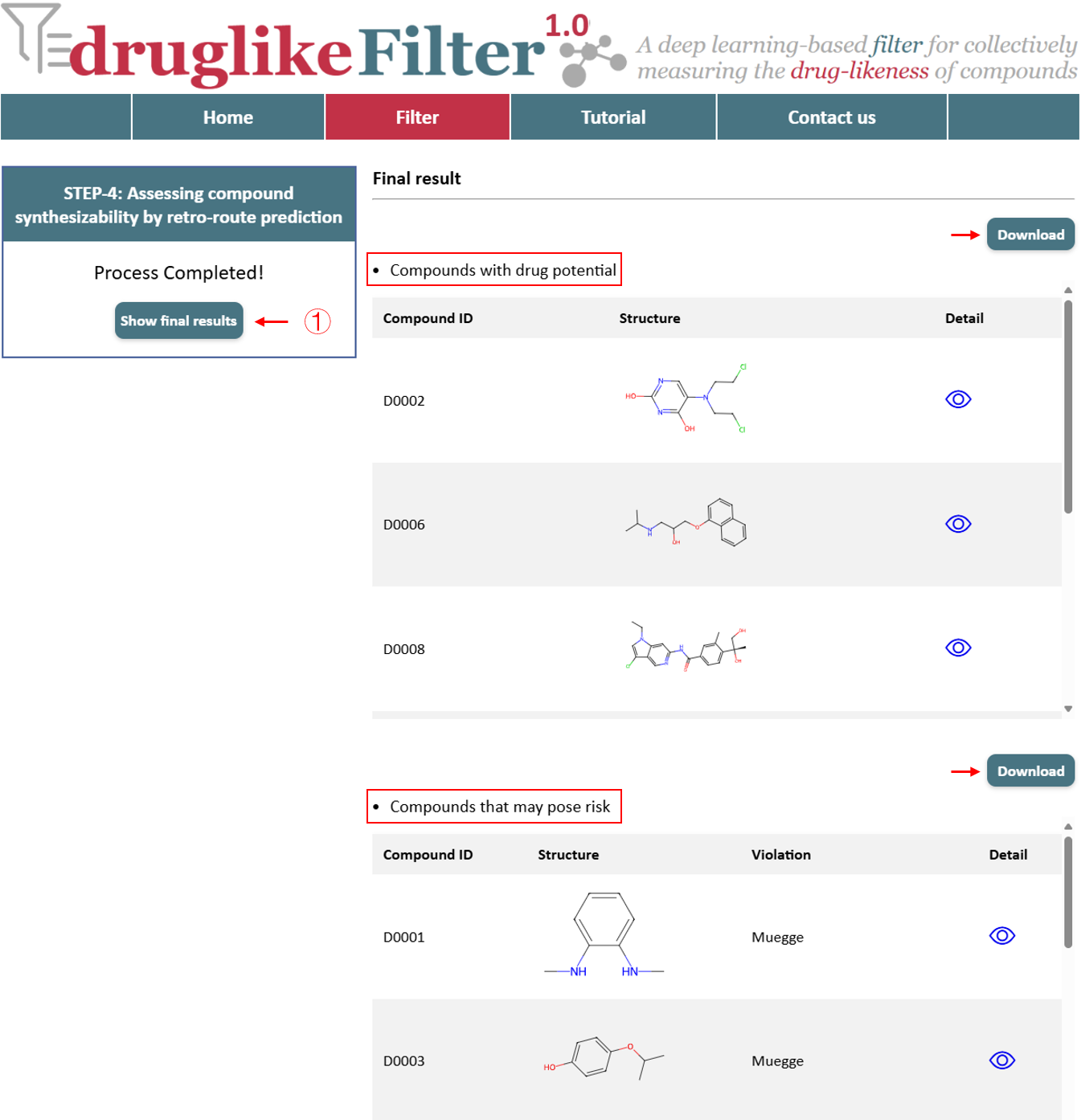
5. Assessing compound synthesizability by retro-route prediction
druglikeFilter offers both an overall synthetic difficulty assessment and retrosynthetic planning to accelerate the chemical synthesis.
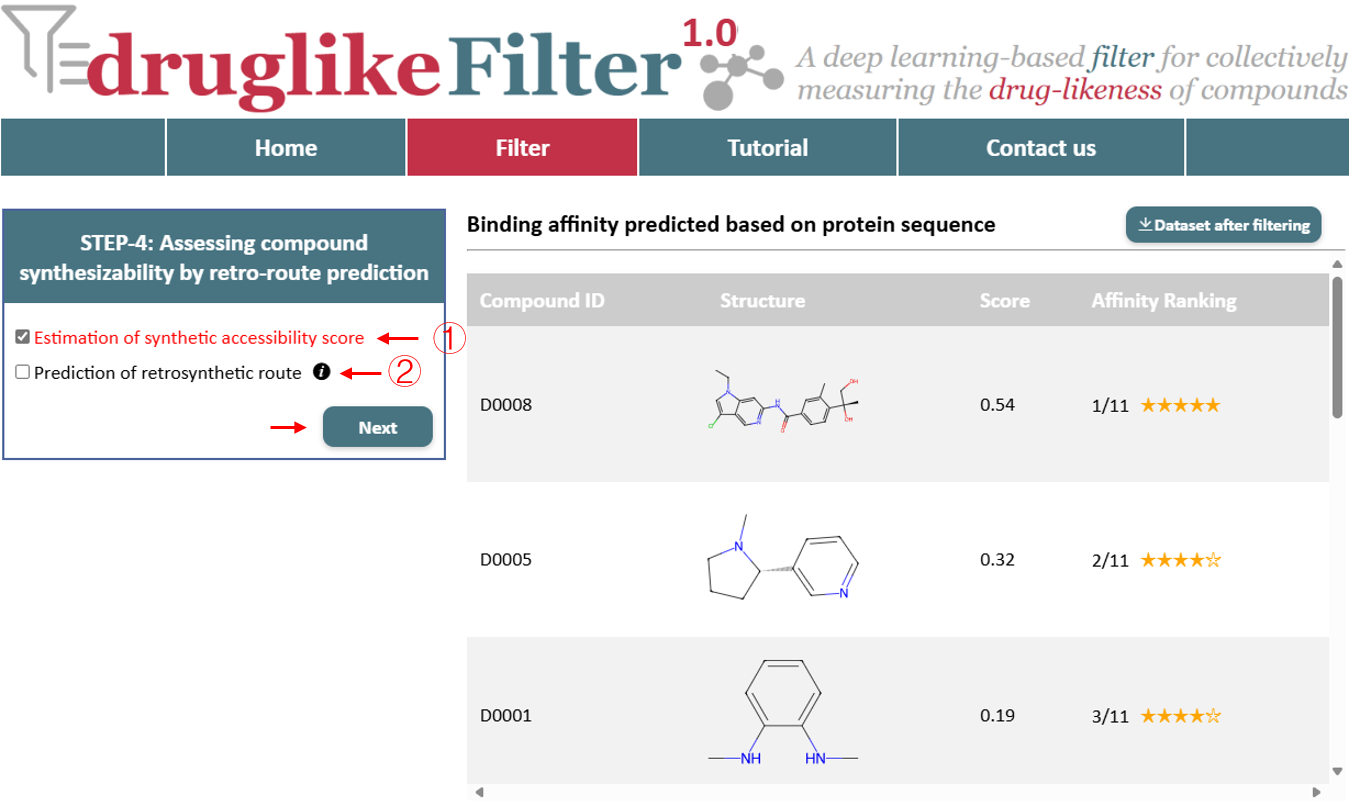
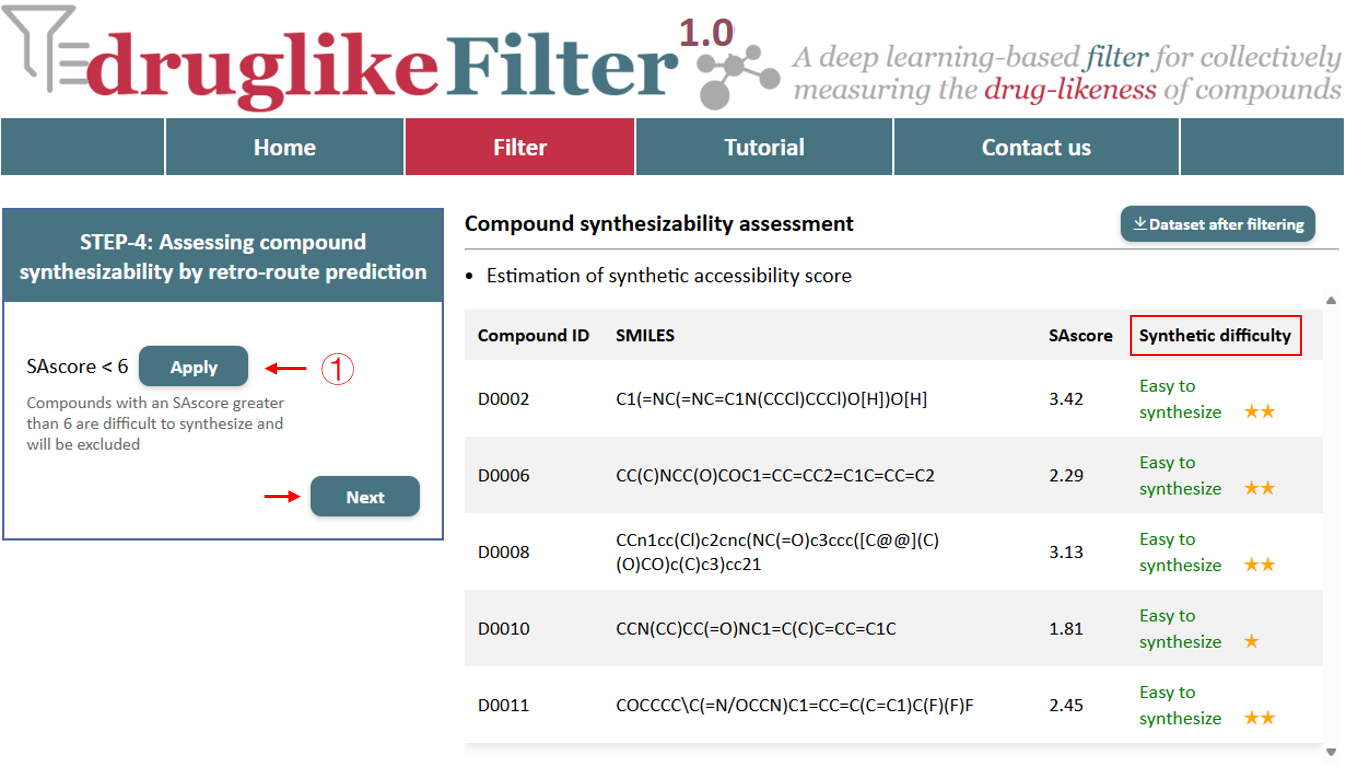
5.1 Estimation of synthetic accessibility score
druglikeFilter first assesses the synthetic difficulty of compounds by calculating the accessibility score (SAscore), which ranges from 1 to 10. A score of 6 or higher indicates a compound is challenging to synthesize, with higher star ratings meaning more challenging.
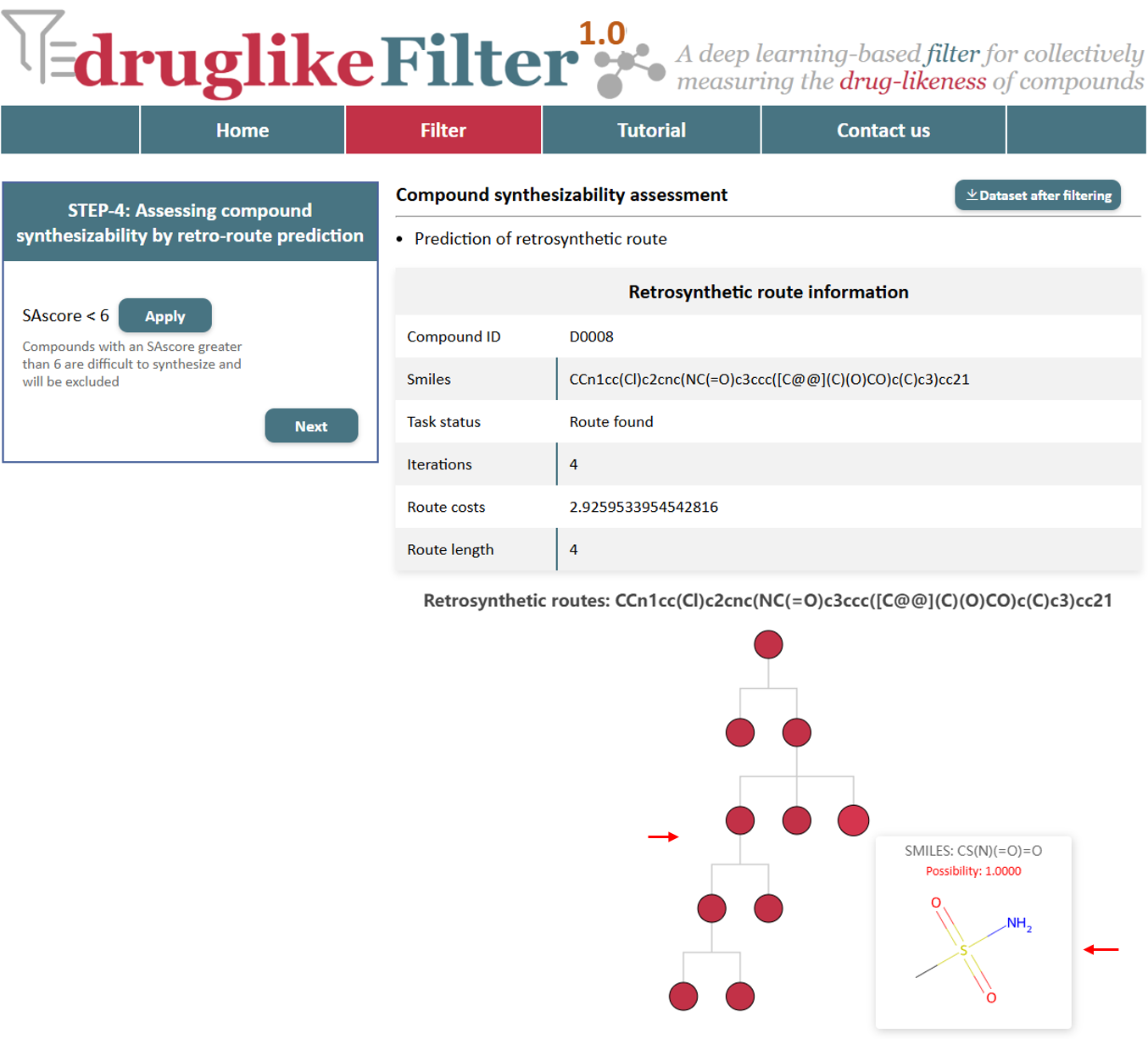
5.2 Prediction of retrosynthetic route
druglikeFilter provides comprehensive retrosynthetic planning for the compound, including reaction routes, synthetic intermediates, reaction feasibility for each step, route cost, and estimated time. The nodes of the tree represent reactants and products and the lines represent reactions. The tree can be expanded and hidden by clicking on the nodes.
Click the “Show final results” button to see all the results. druglikeFilter categorizes the uploaded compounds into two subsets from four dimensions: compounds with drug potential and compounds that may pose iisk. Each subset can be downloaded for further analysis.